MALDI MS/MS biotypization protocol
MALDI PROTOCOL FOR IDENTIFICATION OF MICROORGANISMS BY PROTEIN READING CONCEPT
Introduction
The protein-reading concept using CAF-/CAF+ reagent (chemically activated fragmentation negative/chemically activated fragmentation positive) enables fast, highly accurate, reliable and easy to use identification of microorganisms down to the species level. It is one step derivatization reaction easily incorporated into existing workflows commonly used by biological scientists, as outlined in Figure 1.
SAMPLE PREPARATION (Step 1)
Isolate the proteins from your sample & perform trypsin
digestion
↓
DERIVATIZATION (Step 2)
Add CAF-/CAF+ reagent to peptides
SEPARATION & ANALYSIS (Step 3)
Separate tagged peptides and spot them onto MALDI plate
IDENTIFICATION (Step 4)
Identify microorganism by MALDI-MS/MS & ProteinReader*
Figure 1. Workflow of ESI-MS/MS identification of microorganisms using CAF-/CAF+ reagent.
*ProteinReader is expert software developed exclusively for species identification
CAF-/CAF+ is a chemical reagent for the derivatization of peptide samples prior to analysis by MALDI (Matrix Assisted Laser Desorption Ionization) Tandem Mass Spectrometry (MS/MS). Derivatization procedure of tryptic peptides at N-terminus is illustrated by the following reaction (Figure 2.):
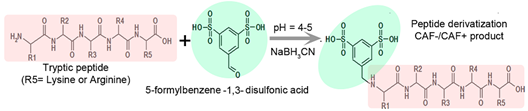
Figure 2. Derivatization procedure of tryptic peptide with CAF-/CAF+ reagent (5-formylbenzene-1,3-disulfonic acid).
For the first time, mass spectrometry can exploit both positive and negative ion mode, either for species or proteins identification.
The CAF-/CAF+ method enables de novo sequencing of derivatized peptides with negative and positive ion mode tandem mass spectrometry (MS/MS– and MS/MS+). Peptide sequences are read from MS/MS spectra and matched against the NCBInr database by developed software named ProteinReader and confirmed by the mass spectrometry data of elucidated peptide mass sequences derived from the annotated genome.
Benefits of using CAF-/CAF+ derivatization reagent
- Accurate
- CAF-/CAF+ is a mild derivatizing reagent with no side reactions resulting in peptide fingerprinting exclusively in negative ion mode (MS–) without interferences and adducts (e.g. sodium, potassium, ammonium) and peptide amino acid sequence analysis in positive and negative MS/MS+/- .
- Fast
- CAF-/CAF+ reagent significantly reduces the derivatization
time to only 10 minutes.
The whole process of sample identification from sample preparation to species identification can be finished in maximum 4 h depending on sample complexity.* - Reliable
- CAF-/CAF+ reagent allows de novo sequencing from spectra of both b-ions (obtained in negative ion mode) and y-ions (obtained in positive ion mode) making sequence reading unambiguous and easy by using two orthogonal techniques (MS/MS negative and MS/MS positive). By reading the same sequence twice forward and backward probability of misreading or misinterpretation of microorganism ID is minimized.
*The protocol consists of two phases: First is sample preparation (protein extraction and digestion) and second is sample treatment & processing (derivatization, separation and identification). Sample preparation can take from one to two hours depending on sample complexity and digestion method used (trypsin tip or trypsin magnetic beads). n. b. overnight in-solution tryptic digestion (9-18 h) can be applied, as well.
Materials, reagents, instruments, software
Materials
Materials enclosed in brackets are the ones used in our laboratory procedures.
- Tissue homogenizer (TissueRuptor and TissueRuptor disposable probes, Quiagen, Germany)
- 2.0 mL PP tubes for cell lysis (Eppendorf, Germany)
- 1.5 mL microcentrifuge tubes (Eppendorf, Germany)
- Tube holder for 1.5 mL tubes
- Tube holder cool pack for 1.5 or 2.0 mL tubes
- Extraction manifold SPE system (Waters, USA)
- SPE 1 mL Diol cartridge, 100 mg Sorbent per Cartridge (Sep-Pak® Diol 1 cc Vac cartridge, Waters, USA)
- Vortexer for mixing
- Laboratory centrifuge with centrifugal force of at least 2,000 rcf (Centric 400, Tehtnica, Slovenia)
- Bench-top microcentrifuge with cooling system and centrifugal force of at least 4,500 rcf (Centrifuge 5415 R, Eppendorf, Germany)
- Vacuum concentrator (Concentrator 5301, Eppendorf, Germany)
- Thermomixer (Thermomixer comfort, Eppendorf, Germany)
- LC Column C18, 300 µm x 150 mm I.D., 3.5 μm particle size (Symmetry 300TM,Waters, USA)
- Spin filters 0.22 µm, cellulose acetate membranes (Agilent)
- Household microwave oven (HeatWave compact 800W, Electrolux)
Reagents used
- Ammonium bicarbonate, ≥ 99.0% (Sigma-Aldrich)
- Triton X-100 (Sigma-Aldrich)
- Mag-Trypsin, trypsin immobilized on magnetic beads (Clontech, USA)
- Acetonitrile, gradient grade for liquid chromatography (Merck, Germany)
- Ammonium formate, for HPLC, ≥ 99.0% (Sigma-Aldrich)
- Trifluoracetic acid, for spectroscopy (Merck, Germany)
- CAF-/CAF+ reagent (5-formylbenzene-1,3-disulfonic acid disodium salt hydrate, p.a. synthetic product, Ruđer Bošković Institute)
- Sodium cyanoborohydride, for synthesis (Merck, Germany) li>Potasium dihydrogen phosphate, p.a. (Kemika, Croatia)
- α-Cyano-4-hydroxycinnamic acid, ≥ 98.0% (TLC) (Sigma-Aldrich)
- Ultrapure water, TOC < 5 ppb, resistivity < 18.2 MΩ cm
Instruments
Instruments enclosed in brackets are the ones used in our laboratory procedures.
- CapLC-MALDI spotting system (Capillary LC, Waters, USA and Tempo™ LC MALDI Spotting System, Applied Biosystems, MDS Sciex, USA)
- MALDI TOF/TOF mass spectrometer (4800 MALDI TOF/TOF™ Analyzer, Applied Biosystems, MDS Sciex, USA)
Software
- Protein Reader (software developed by “Ruđer Bošković” Institute and Faculty of Food Technology and Biotechnology, University of Zagreb)
Procedure
STEP 1 – SAMPLE PREPARATION
Removing cell suspension growth medium
Centrifuge suspension of cells in 50 or 15 mL culturing tubes 2,000 rcf for 15 min and aspirate the supernatant. Resuspend the pellet in 1.5 mL of 25 mM NH4HCO3, transfer to a 2.0 mL tube and centrifuge at 4,500 rcf for 20 min at 4°C. Aspirate the supernatant. Repeat 2-4 more times, depending on the sample.
Cell lysis
Add 400 µL of cell lysis buffer (25 mM NH4HCO3 + 0.1%
Triton-X-100) and resuspend the pellet. Keep the tubes incubated
on cool pack.
Proceed to cell rupture using TissueRuptor and disposable probe
with the following settings:
Power: Position II
Grinding cycle: 45 seconds ON / 45 seconds OFF
Total grinding times: 5-6 cycles
Blocking endoprotease activity
Stop the TissueRuptor after 5 cycles, close the lid on 2.0 mL tube and put the sample in boiling water for 3 minutes to inhibit endoprotease activity.
Obtaining solution of proteins
Remove the sample tube from the water bath and centrifuge at 4,500 rcf for 20 min at 4°C to remove disrupted cell material. Transfer the supernatant containing soluble proteins to a clean 1.5 mL tube.
Protein digestion
Use one of the three trypsin digestion procedures:
- Mag-Trypsin (trypsin immobilized on magnetic beads)
- Trypsin tip
- Standard in-solution tryptic digestion
STEP 2 – DERIVATIZATION
Derivatization procedure
Reconstitute each sample containing the evaporated tryptic peptide mixture with 60 µL of derivatization solution. The derivatization solution contains 12.5 mM 5-formylbenzene-1,3-disulfonic acid disodium salt hydrate (p.a. synthetic product, Ruđer Bošković Institute) and 95.5 mM of NaBH3CN dissolved in 10 mM KH2PO4 and adjusted to pH 5. Close the tube with the sample, place it in the holder (e.g. styrofoam) and perform the derivatization in a household microwave oven at 90W for 10 minutes.
STEP 3 – SEPARATION
Solid Phase Extraction (SPE) – optional
Purify the peptides after trypsin digestion on Diol cartridges using vacuum manifold SPE system. Condition the Diol cartridges in three steps: first, aspirate and dispense to waste three times 0.5 mL of 80% acetonitrile (ACN) mixed with 20% water solution of 0.1% ammonium formate (AF) (v/v); second, aspirate and dispense to waste three times 0.5 mL of 50% ACN mixed with 50% water solution of 0.1% AF (v/v); and finally, aspirate and dispense to waste three times 0.5 mL of an aqueous solution of 0.1% AF (v/v). Load the peptide sample solution onto the cartridge and wash five times with 0.5 mL aqueous solution of 0.1% AF (v/v). Elute the peptides from the column to a clean 1.5 mL tube with 0.35 mL of 80% ACN mixed with 20% aqueous solution of 0.2% AF (v/v). Evaporate the eluting solution to dryness in vacuum centrifuge
Sample Filtration
After derivatization, filter the sample before loading LC column by passing it through cellulose acetate spin filter 0.22 µm using low-speed centrifugation (e.g at 4000 rpm for 30 s).
Separation by Capillary Liquid Chromatography
The CapLC system equipped with a Photodiode Array (PDA) detector coupled to TempoTM LC MALDI spotter is used for peptide separation and collection directly onto the MALDI plate. Perform the chromatographic separation on a silica based LC column C18, 300 µm x 150 mm I.D., 3.5 μm particle size at 30°C. Set the flow rate to 2 µL/min and injection volume to 8 μL. Vial temperature is maintained at 5°C in the autosampler tray. Use following mobile phases: mobile phase A consists of 0.1% TFA aqueous solution and mobile phase B consists of 80% ACN mixed with 20% aqueous solution of 0.1% TFA (v/v). Eluted derivatized peptides are detected by UV absorbance at 280 nm. Program the 50 min gradient elution to increase the percentage of solvent B from 5% to 80% over 35 min and then to condition the column back to the initial conditions until completion of the run. Set complete gradient conditions following the Table 1.
Table 1. Complete gradient conditions
| Time (min) | A (%) | B (%) |
|---|---|---|
| 0 | 95 | 5 |
| 7 | 95 | 5 |
| 35 | 20 | 80 |
| 40 | 95 | 5 |
| 50 | 95 | 5 |
Set the spotter make-up flow to 2 μL/min (5 mg α-Cyano-4-hydroxycinnamic acid matrix dissolved in 1 mL of 50% ACN aqueous solution). Example of typical LC-MALDI chromatogram is shown in Figure 3.
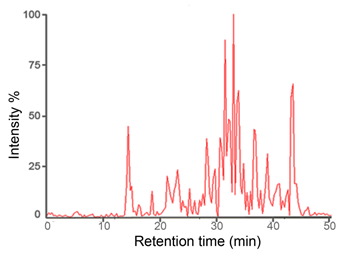
Figure 3. Example of typical LC-MALDI chromatogram.
STEP 4 – IDENTIFICATION
Mass spectrometry
MS acquisition (CAF-/CAF+) is performed with a MALDI TOF/TOF 4800 Plus analyzer equipped with a 200 Hz, 355 nm neodymium-doped yttrium aluminum garnet Nd:YAG laser. Ions are analyzed in reflectron negative ion mode. The instrument parameters are set using the 4,000 Series Explorer software version (V 3.5.3, Applied Biosystems, USA). Mass spectra are obtained by averaging 1,800 laser shots covering a mass range of m/z 1,000 to 4,000.
MS/MS acquisition is achieved by 1kV collision energy, first in negative than in positive ion mode without usage of collision gas. The same precursor ions generated by negative ion MS were analyzed in negative and positive MS/MS.
Precursor peak selection
LC-MALDI peak MS processing was controlled by so called job-wide peak selection method. Peak selection was done automatically in order to analyze the MS spectra and acquire MS/MS data from selected peaks. Parameters of MS/MS peak selection were as follows: Minimum S/N filter 15, Minimum chromatogram peak width 3 (used to prevent redundant peak analysis in adjacent fractions), Maximum precursors per fraction 20, Fraction-to-fraction precursor mass tolerance 200 ppm.
ProteinReader identification
By depleted MS/MS spectra derived out of derivatized peptides (only b-ions in negative ion mode and y-ions in positive ion mode are observed in MS/MS) peptide sequences can be read by specific software named ProteinReader (Figure 4).
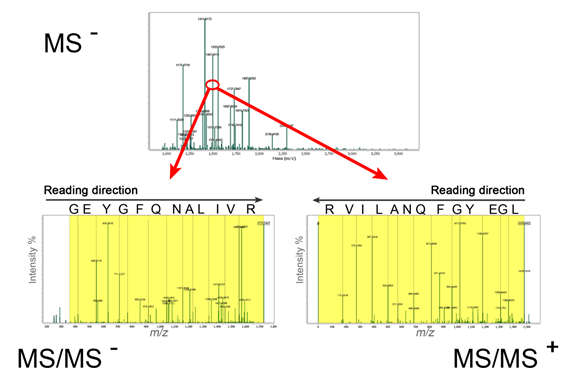
Figure 4. De novo sequencing of derivatized peptide LGWYGFQNALIVR, m/z 1497.6111 obtained in MS– and sequenced in MS/MS– and MS/MS+ by ProteinReader software.